
What is Senescence and How Does it Relate to Cancer?
“Senescence is a cellular response to a variety of stress signals that is characterized by a stable withdrawal from the cell cycle and major changes in cell morphology and physiology. While most research on senescence has been performed on non-cancer cells, it is evident that cancer cells can also mount a senescence response. In this Review, we discuss how senescence can be induced in cancer cells. We describe the distinctive features of senescent cancer cells and how these changes in cellular physiology might be exploited for the selective eradication of these cells (senolysis). We discuss activation of the host immune system as a particularly attractive way to clear senescent cancer cells. Finally, we consider the challenges and opportunities provided by a ‘one-two punch’ sequential treatment of cancer with pro-senescence therapy followed by senolytic therapy.
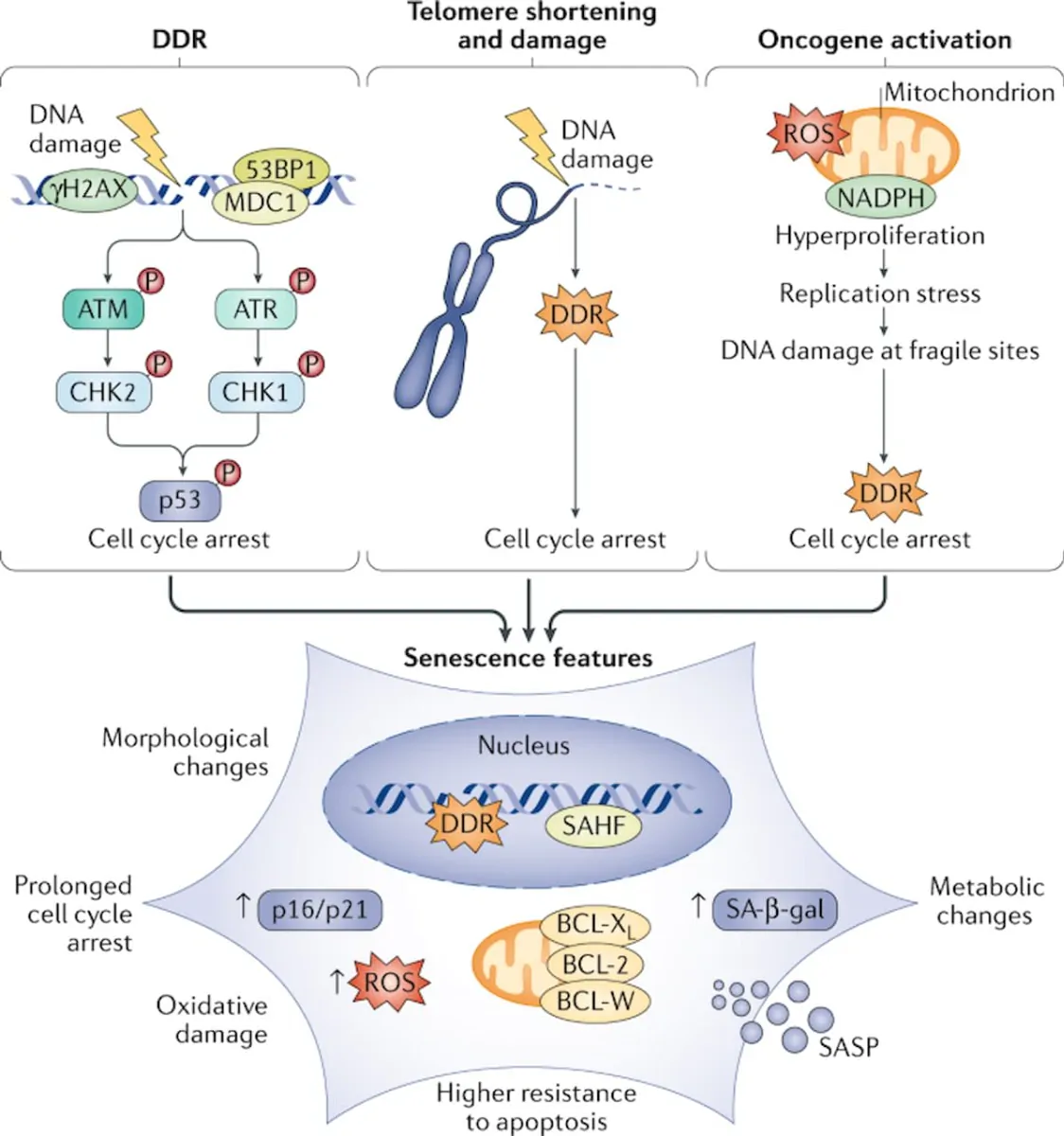
Cells progressively accumulate damage during their lifetime due to a multitude of endogenous and exogenous stressors. Although an array of repair mechanisms exists, the damage can sometimes be persistent. Replicative cells also require defence mechanisms to reduce the impact of these persistent stresses on tissue degeneration and to restrict cancer development. One of these defence mechanisms involves switching to a stable non-proliferative, metabolically active survival state named cellular senescence. Cellular senescence can be induced by a variety of stress and damage signals. The phenomenon was first described in 1961 by Hayflick and Moorhead, who observed that replicative cells have a limited number of divisions — the ‘Hayflick limit’ — that we now understand to be caused by telomere shortening; this is sensed by the cells as a persistent DNA damage response. As a consequence, these cells undergo senescence to avoid further genomic instability and accumulation of DNA damage. In addition to telomere shortening, various other stress stimuli can trigger senescence, including oncogenic stress, oxidative stress, lysosomal or endoplasmic reticulum stress, nutrient depletion, and genotoxic stress induced by cancer therapies or even from pathogens, including coronavirus. Induction of senescence in response to stress stimuli other than telomere attrition is sometimes referred to as stress-induced premature senescence.” (1)
(2)
“Induction of senescence in response to stress stimuli results in stable cell cycle arrest. Therefore, senescence represents a defence mechanism against cancer. However, cell cycle withdrawal is also a feature of several other cell states such as quiescence, terminal differentiation, dormancy or drug-tolerant persistence. It is difficult to unambiguously differentiate between these related cellular states, mainly because we lack ‘gold standard’ markers of the senescent state. Heterochromatinization of proliferative genes is often seen in senescent cells, and this is thought to enforce a durable cell cycle arrest that cannot be readily reversed by pro-mitogenic stimuli. However, as epigenetic modifications are reversible in principle, it is probably incorrect to consider senescence as a strictly irreversible arrest. Indeed, several studies have highlighted the reversible nature of senescence. In addition to resistance to proliferative stimuli, another hallmark of senescent cells is resistance to cell death via resistance to apoptotic stimuli or upregulation of pro-survival pathways. In this way, senescent cells remain in a viable state. Apoptotic resistance is regulated by persistent activation of anti-apoptotic proteins, such as the BCL-2 protein family, as well as epigenetic repression of pro-apoptotic proteins such as BAX. Senescent cells are often also characterized by increased lysosomal capacity. This is linked to increased activity of the lysosomal enzyme senescence-associated β-galactosidase (SA-β-gal), which is one of the most widely used markers for senescence although it is neither required nor specific for the senescence phenotype. Other hallmarks of senescence include altered cell metabolism by mitochondrial dysfunction, macromolecular damage, endoplasmic reticulum stress, loss of lamin B1 (a protein of the nuclear lamina), and the overexpression or activation of cell cycle inhibitors INK4A and ARF (both encoded by CDKN2A), p21 (encoded by CDKN1A) and p53 (encoded by TP53).” (1)
Inducing Senescence in Cancer Cells.
“A substantial number of anticancer interventions induce senescence in cancer cells by triggering genotoxic stress, hyperactivation of mitogenic signalling or oxidative stress, leading to stable cell cycle arrest and SASP induction. Therefore, therapy-induced senescence serves as an initial antitumour mechanism to halt proliferation and prevent further genomic instability (Fig. 1). “
“Low doses of chemotherapy particularly trigger a senescent cell state in human cancer cells, while apoptosis is induced at higher doses.
This finding might explain why often only a subset of cancer cells become senescent in response to conventional chemotherapies or radiotherapies as the senescence response is only triggered in a specific window of DNA damage. Mechanistically, many chemotherapies cause DNA damage in cancer cells, which triggers senescence through ATM–CHK2 and ATR–CHK1 kinase-mediated activation of the interconnected p53–RB pathways (Fig. 1). Topoisomerase I and II inhibitors, such as doxorubicin, etoposide and camptothecin, are widely used for the treatment of a variety of cancer types and have been shown to dysregulate re-ligation of DNA strands after supercoil unwinding. This leads to massive DNA damage and increased expression of p53 and its downstream targets CDKN1A and PAI1 (also known as SERPINE1), subsequently inducing senescence. Platinum-based compounds, such as cisplatin, carboplatin and oxaliplatin, also induce extensive DNA damage through DNA cross-linking, resulting in senescence induction. Similarly, alkylating agents, such as temozolomide, dacarbazine and busulfan, form DNA crosslinks by reacting with atoms in DNA, triggering a DNA damage-mediated senescence response. Microtubule inhibitors, such as paclitaxel, docetaxel and vinca alkaloids, dysregulate the normal microtubule spindle dynamics to impair metaphase–anaphase transition and arrest the cells at mitosis. This cell cycle dysfunction may also cause extensive DNA damage and trigger a p53–p21-facilitated senescence response. Methotrexate and gemcitabine both induce genotoxic stress by blocking DNA synthesis, thereby inducing cellular senescence. It is important to keep in mind that, while quite a few existing chemotherapeutics have some ability to induce senescence, the apoptotic response is dominant in most cancers. As such, most chemotherapies are unable to induce senescence in a significant fraction of cancer cells in vivo.” (1)
Killing Senescent Cancer Cells.
“Selective killing of senescent cells — senolytic therapy — has initially been developed to reduce age-related symptoms and improve healthy longevity but senolytic therapy may also have utility for the eradication of senescent cancer cells.”
“Compounds that modulate p53 activity can also act as senolytics. For example, the peptide FOXO4-DRI interferes with the interaction between p53 and FOXO4, resulting in p53 nuclear exclusion followed by apoptosis selectively in senescent non-cancer cells (Fig. 3e). In mice, the FOXO4-DRI peptide was well tolerated, improved health span and neutralized doxorubicin-induced chemotoxicity caused by therapy-induced senescent cell burden. Although this peptide demonstrates promising senolytic activity in mice, its therapeutic use still needs to be clinically evaluated. The senolytic compound UBX0101 also modulates p53 activity... Notably, disruption of the p53–MDM2 complex could also induce apoptosis instead of senescence as demonstrated by the senolytic drug UBX0101 or the forkhead box protein O4 (FOXO4)-DRI peptide in non-cancer models, which is discussed in more detail below. Whether this approach will also work for cancer has yet to be investigated (Fig. 1).” (1)
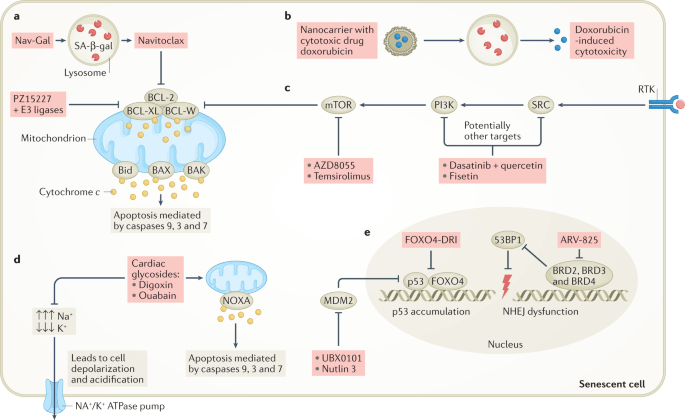
“FOXO4 is a transcription factor that plays a role in the maintenance of senescent cell viability. In addition, its presence in a small fraction of nonsenescent adult cells makes it a potential target to eliminate senescent cells. FOXO4 binds p53 and prevents it from inducing the apoptosis of senescent cells. For this reason, impairing this FOXO4-TP53 interaction may be useful to activate apoptosis in these cells. FOXO4-DRI is a cell-permeable peptide that includes part of the p53-interaction domain of FOXO. This peptide could compete with endogenous FOXO4 for p53, impairing the FOXO4-p53 interaction, and inducing apoptosis in senescent cells. Several studies have shown that FOXO4-DRI selectively eliminates senescent cells, without affecting nonsenescent ones. Another peptide recently developed by molecular modelling is ES2, which has the same function as the previously described. The authors demonstrated that combination therapy of ES2 and a Braf inhibitor results in apoptosis and a survival advantage in mouse models of Braf mutant melanoma and reduced senescent cells in ageing mice. Furthermore, they showed that ES2 is effective at eliminating both normal and cancer senescent cells.” (3)
“In another study, human cancer associated fibroblasts (CAFs) were treated with x-rays and showed a senescence phenotype, which could promote cancer cell growth via the JAK/STAT signaling pathway. The same study found that FOXO4-DRI, a FOXO4-p53–interfering peptide, could kill CAFs and sensitize tumor cells to radiation treatment (Meng et al., 2021).” (4)
Inflammatory SASP Secreted by Senescent Cells are a Double-Edged Sword in Cancer.
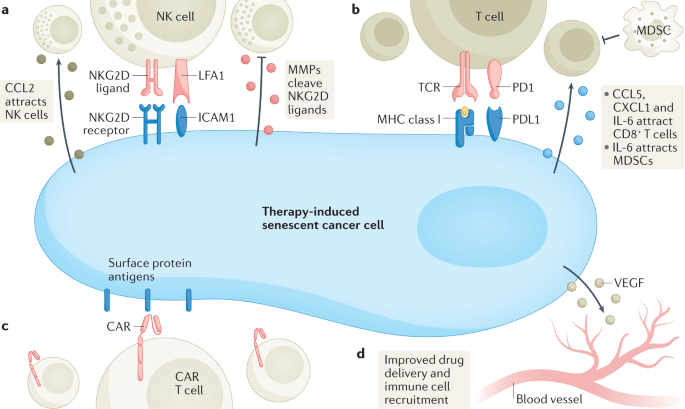
“Through their SASP, senescent cancer cells can also arrest neighbouring cancer cells, improve the vasculature for drug delivery and recruit immune cells that can contribute further to tumour suppression. However, persistence of therapy-induced senescent cells may be detrimental in the long term by creating a pro-inflammatory, immunosuppressive microenvironment. Moreover, the SASP can promote angiogenesis to advance tumour growth and an epithelial to mesenchymal transition in neighbouring cancer cells, which can enhance migration and promote metastasis.”
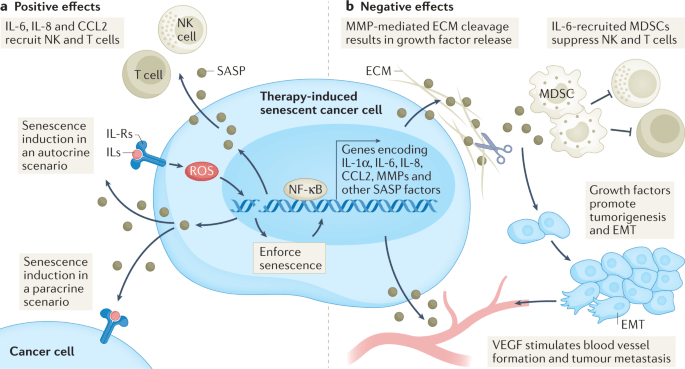
“Arguably, one of the more distinctive, yet not exclusive, features of senescence is the secretion of pro-inflammatory cytokines, growth factors and matrix metalloproteinases (MMPs), collectively termed the senescence-associated secretory phenotype (SASP). The first evidence of a SASP produced by senescent cells was a gene expression study performed in 1999 demonstrating the expression of inflammation-associated genes in senescent fibroblasts, although the term ‘SASP’ was not used until 2008. The SASP is generated as a result of activation of the nuclear factor κB (NF-κB), cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING), GATA4 (ref.), CCAAT/enhancer-inding protein-βa (CEBPβ), NOTCH, IL-6, Janus kinase (JAK)–signal transducer and activator of transcription (STAT), p38 MAPK and mTOR pathways. The SASP factors can reinforce senescence in an autocrine fashion and influence the tissue microenvironment by paracrine signalling to adjacent tumour, non-tumour and immune cells. Once the levels of senescent cells achieve a certain threshold, they can even produce systemic effects, causing various ageing-related diseases.
A substantial number of anticancer interventions induce senescence in cancer cells by triggering genotoxic stress, hyperactivation of mitogenic signalling or oxidative stress, leading to stable cell cycle arrest and SASP induction. Therefore, therapy-induced senescence serves as an initial antitumour mechanism to halt proliferation and prevent further genomic instability (Fig.1). Through their SASP, senescent cancer cells can also arrest neighbouring cancer cells, improve the vasculature for drug delivery and recruit immune cells that can contribute further to tumour suppression. However, persistence of therapy-induced senescent cells may be detrimental in the long term by creating a pro-inflammatory, immunosuppressive microenvironment. Moreover, the SASP can promote angiogenesis to advance tumour growth and an epithelial to mesenchymal transition in neighbouring cancer cells, which can enhance migration and promote metastasis.” (1)
References
1. Wang, Liqin, et al. “Exploiting Senescence for the Treatment of Cancer.” Nature Reviews Cancer, 3 Mar. 2022, 10.1038/s41568-022-00450-9.
2. Di Micco, Raffaella, et al. “Cellular Senescence in Ageing: From Mechanisms to Therapeutic Opportunities.” Nature Reviews Molecular Cell Biology, vol. 22, no. 2, 16 Dec. 2020, pp. 75–95, 10.1038/s41580-020-00314-w.
Product available for research use only.

